SIMULATION OF GENE EVOLUTION (evidence for GC-NSF(a) hypothesis on the origin of genes)
Kenji Ikehara
Kita-uoya-nishi-machi, Nara 630-8506, Japan
Fax/phone: +81-742-20-3402; E-mail: ikehara@cc.nara-wu.ac.jp
(Received 3 April 2003, Accepted 27 August 2003)
Abstract
We have previously proposed a hypothesis on the origin of genes, suggesting that genes generally originated from nonstop frames on antisense strands of GC-rich genes (GC-NSF(a)s) under the universal genetic code [Ikehara, K., et al., Nucl. Acids Res., 24, 4249 (1996)]. To obtain evidence for the hypothesis, simulation of gene evolution was carried out under the six conditions for folding of polypeptide chains into appropriate three-dimensional structures using a Mycobacterium tuberculosis GC-NSF(a) (508 codons) as an ancestor gene. The results showed that the simulation well reproduces both the base compositions at three codon positions and the average amino acid compositions of extant proteins encoded by microbial genomes when a conserved region (200 codons) in simulated proteins was set at about 40%. Contrary to that, the guanine composition at the first codon position and the average amino acid compositions considerably deviated from those of extant genes and proteins when the simulation was carried out using a Borrelia burgdorferi AT-rich gene as an ancestor gene in the presence or the absence of about 40% conserved regions. These results apparently support the GC-NSF(a) hypothesis on the origin of genes which we have proposed.Key Words: GC-NSF(a) hypothesis, origin of genes, computer simulation, gene evolution, protein evolution
Introduction
Since the first complete microbial genomic sequence of Haemophilus influenzae was published in 1995 by the Institute for Genomic Research (TIGR) [1], about 100 microbial genomes have been sequenced and published [2]. In the past several years, even the base sequences of eukaryotic genomes, such as a fungus (Saccharomyces cerevisiae), a plant (Arabidopsis thaliana), a nematode (Cenorhabditis elegance), and a human (Homo sapiens), were determined and published [2]. In parallel with the determination of the genomic sequences, information about amino acid sequences of proteins has been rapidly accumulated. Despite the recent advances in the understanding of many genetic functions and bacterial genomes, the kind of base sequences of the very first genes remains largely unknown. In addition, what has determined the respective base compositions at the codon position and average amino acid compositions in bacterial proteins (Fig. 1) is also unknown. One of the keys for solving these riddles would be to know from what kind of sequences extant genes originated and how the ancestor genes evolved.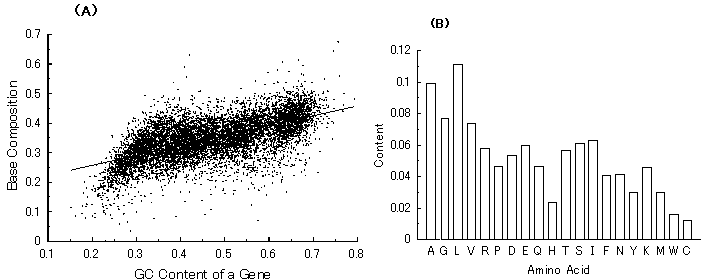
Fig. 1. (A) Guanine composition at the first codon position of genes from 7 microbial genomes (Mycobacterium tuberculosis, Aeropyrum pernix, Escherichia coli, Bacillus subtilis, Haemophilus influenzae, Methanococcus genitarium and Borrelia burgdorferi). (B) Amino acid composition of E. coli proteins (1,525) encoded by the E. coli genome. The number in parenthesis represents the protein number analyzed. Capital letters below a horizontal axis show amino acids written with one-letter symbols.
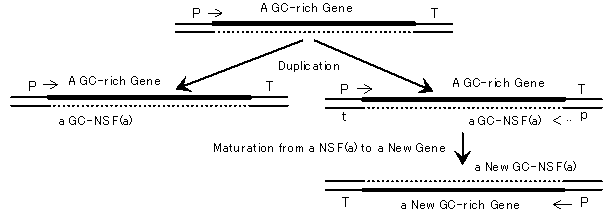
Fig. 2. A GC-NSF(a) hypothesis on the origin of genes, predicting that new genes originate from nonstop frames on antisense strands of GC-rich genes (GC-NSF(a)s), which appear at a high probability. I supposed that a GC-rich NSF(a) gene would evolve to get a larger activity by accumulating necessary mutations when a protein was expressed from a promoter (p) and the GC-NSF(a) possessed even the faintest activity necessary for life. Then, the new GC-rich gene could be propagated to other bacteria having more AT-rich genes under uni-directional AT-mutation pressure (gene evolution). Capital letters, P and T, and small letters, p and t, mean functional promoters and effective transcription terminators, and latent promoters and inefficient terminators, respectively. Thick lines and dotted lines indicate active genes and cryptic genes, respectively.
Computational Methods
Collection of base sequences of bacterial gene. Hypothetical GC-NSF(a) genes, which appeared on antisense strands of Mycobacterium tuberculosis Rv0001 (dnaA; 508 codons), Rv0003 (recF; 385 codons), Rv0062 (celA; 380 codons) and Rv0074 (unknown function; 411 codons), were used in simulation of gene evolution. Those genes were collected from a data-bank registered in Anonymous FTP of the GenomeNet in Bioinformatics Center, Institute for Chemical Research, Kyoto University. A Borrelia burgdorferi AT-rich gene (BB0024 (unknown function; 379 codons)) used as a control gene for the simulation was also obtained from the data-bank. Strategy for simulation of gene evolution. Simulations were carried out using a handy-made program written in C-language (Fig. 3). For the simulation, a nucleotide sequence on antisense strand of a GC-rich M. tuberculosis Rv0001 gene was mainly used as a hypothetical ancestor gene (GC-NSF(a)). Stop codons which appeared in the frame of the GC-NSF(a) were exchanged to sense codons by changing the first base T to G before the simulation. Before practicing the simulation, it had been confirmed that the hypothetical ancestor protein satisfied the six conditions (hydropathy index, indexes for secondary structure formations (a-helix, b-sheet and b-turn), acidic amino acid and basic amino acid contents) for folding a polypeptide chain into an appropriate three-dimensional structure in water [6].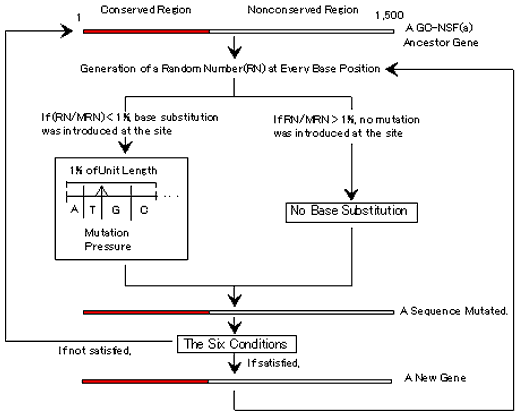
Fig. 3. Strategy of simulation of gene evolution from a hypothetical ancestor gene, GC-NSF(a) with 1,500 bases (500 codons) (see also Computational Methods). A random number (RN) was generated at every base position and original base was substituted to either of four bases when the random number was below 1% of the maximum random number (MRN). A base to be replaced was determined by the four fractions where the random number was located. When a hypothetical protein encoded by a mutated gene satisfied the six conditions, the simulation procedure was repeated using the base-substituted sequence. These procedures were repeated until getting a total of 1,000 hypothetical functional genes. AT-mutation pressure was changed step-wise 5% by 5% from 75 to 25%. A conserved region was set on the ancestor gene, to be remained invariable of amino acids in a region from the N-terminal end to an amino acid at a fixed position, even if bases in the regions were altered.
The structural indexes of a whole protein were calculated from the amino acid composition in a protein and the six structural indexes of amino acids [6], which was described in Stryer's text book [7]. The conditions for selection of mutated genes during simulation of gene evolution were set up based on the average values of the six structural indexes of extant microbial proteins and the respective standard deviations [6]. The average values and the standard deviations are as follows: hydropathy index (-1.513, 0.382); a-helix index (1.027, 0.034); b-sheet index (1.004, 0.023); b-turn index (0.964, 0.045); acidic amino acid content (0.12, 0.032); and basic amino acid content (0.141, 0.031). The restricted ranges for the selection were gradually reduced from the average values plus and minus five-thirds of the respective standard deviations of the first ancestor gene to the average values plus and minus one-third of the standard deviations of the 250th mutated gene.
A random number (RN) was generated in a computer at every base position of the ancestor gene and an original base was substituted to either of four bases when the random number was below 1% of the maximum random number (MRN) (Fig. 3). A base to be replaced was determined by the four fractions where the random number was located, as follows. First, the 1% region was cut into two parts by a fixed AT-mutation pressure determining a ratio of (G+C) versus (A+T). Then, the two parts were subdivided into two halves or into four fractions for G, C, A and T. Next, I examined whether a hypothetical protein encoded by a new base sequence, which was obtained after introduction of substitutions at a probability of 1% at every position of the base sequence, satisfied the six conditions or not. The mutated sequence was removed from the simulation as an inactive gene if one or more number of structural indexes of the simulated protein did not satisfy the respective conditions, or the sequence met any stop codon in a frame of translation. Then, the procedure for introduction of mutations was repeated from one step before. If a simulated protein satisfied all of the six conditions, the simulation of gene evolution was carried out using the mutated sequence as a new ancestor gene. These procedures were repeated under the same AT-mutation pressure until 1,000 hypothetical genes satisfying the six conditions were obtained. The AT-mutation pressure determining GC content was changed from 75 to 25, 5% by 5% step-wise. As a result of this treatment, the base compositions of mutated genes gradually changed from GC-rich to AT-rich.
When a conserved region must be established on an ancestor gene, amino acids in a region from the N-terminal end to an amino acid at a fixed position were kept to the original amino acids upon introduction of base substitutions (Fig. 3). Therefore, a hypothetical gene was allowed to evolve to a new gene when all amino acids in the fixed region were invariable, even if bases in the region were replaced by other bases.
Amino acid composition of hypothetical protein selected independently of gene simulation. To confirm that average amino acid compositions of extant microbial proteins have not been determined simply by the characteristics of 20 natural amino acids, amino acid composition of a hypothetical protein was generated with 20 random numbers in a computer, independently of an ancestor gene and of its evolutionary pathway. Next, if the protein specified by the 20 random numbers satisfied the six conditions for globular protein formation, the protein was selected out as a functional protein. The average values plus and minus one-third of the respective standard deviations were used as restricted ranges for the selection. These procedures were repeated until obtaining 150 hypothetical proteins satisfying the six conditions.
Results and Discussion
GC-NSF(a) Hypothesis on the Origin of Genes. We hypothesized that a GC-rich NSF(a) (an ancestor gene) could evolve to a new gene with a large activity by accumulating necessary base substitutions (gene maturation) if a small amount of protein was expressed from the ancestor gene and the expressed protein possessed even the faintest activity for a required chemical reaction (emergence of a new gene) (Fig. 2). Then, the newly-born GC-rich gene could be propagated to other bacteria having more AT-rich genomes under uni-directional AT-mutation pressure (gene evolution) [3, 4].However, it is very difficult to solve a problem on the origin of something, such as genes, since we must consider events which took place in the past, and past events cannot be reproduced in a laboratory. In such a situation, a powerful approach is a computer simulation of past events. The availability of complete bacterial genome sequences carrying a variety of GC contents has given us a great opportunity to investigate many fundamental questions hidden in living systems. From an analysis of extant proteins, we have confirmed that the six structural indexes (hydrophobicity/hydrophilicity, a-helix, b-sheet and b-turn formabilities, acidic amino acid and basic amino acid contents) are unaffected by the change of GC content of a gene [6]. These properties are especially important, because they are a necessary condition for a simulation of gene evolution. Thus, we actually carried out the simulation in order to find a possible evolutionary process from an ancestor gene (a GC-NSF(a)) to other progeny genes, as described below.
Computer Simulation of Gene Evolution. According to the GC-NSF(a) hypothesis on the origin of genes, it is assumed that many extant genes originated from GC-NSF(a)s on GC-rich genes (Fig. 2). It is also assumed that the GC-rich ancestor genes decreased their GC contents as they accumulated mutations on the nucleotide sequences under uni-directional AT mutation pressure [4]. Then, all of the six indexes of the hypothetical proteins which appeared during the procedures of the gene simulation must be included in the restricted regions required to form appropriate three-dimensional structures (see Computational Methods in more detail).
Simulation of gene evolution was carried out using an antisense sequence on a GC-rich M. tuberculosis gene (Rv0001; dnaA) (a GC-NSF(a)), which is composed of 1,524 bases (508 codons). Six termination codons observed on a GC-NSF(a) of the gene were replaced by other sense codons by changing the first letter T in the codons to G. Mutations were introduced onto the sequences at a probability of 1% at every base position. Firstly, the simulation was carried out under eleven different mutation pressures without setting any conserved region on the ancestor gene. Twelve base compositions dependent on GC content of a hypothetical gene were obtained from the simulation, because 4 bases (A, T, G and C) are used at each codon position. Solid lines, which were obtained by the least square approximation of the base compositions of seven actual microbial genomes, are also drawn in the figure to compare them with the simulated results [4]. The simulation well reproduced the base compositions at three codon positions of actual genes in seven microbial genomes except T1, C3, A3 and G1 (Fig. 4). The alphabetical letter and number indicate the base and the codon position, respectively.
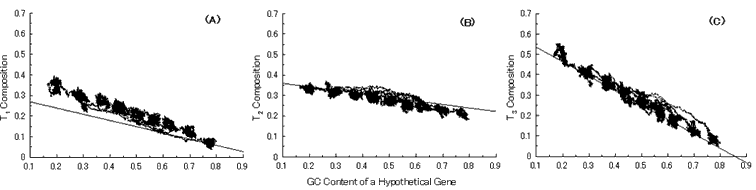
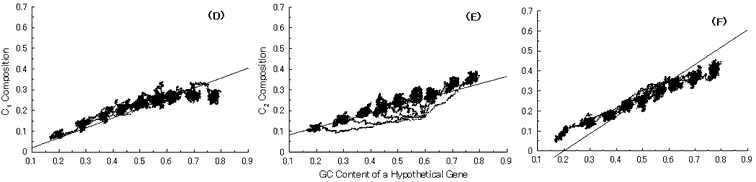
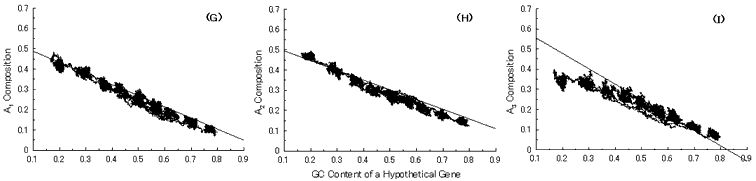
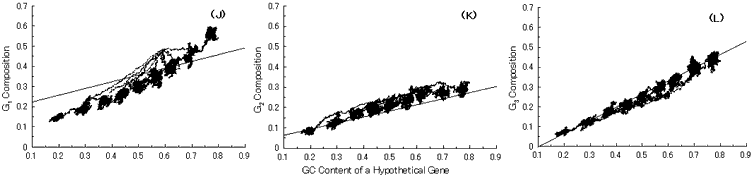
Fig. 4. Simulation of gene evolution from a hypothetical ancestor gene, GC-NSF(a), on M. tuberculosis Rv0001 gene (508 codons) without setting any conserved region in the ancestor protein. Linear lines in the figures represent the average values approximated with the least square method of base compositions at the codon positions of extant genes on seven microbial genomes [6]. Figures (A), (B) and (C) show the dependencies of thymine composition on GC content of a hypothetical gene at the first, second and third codon positions, respectively. Similarly, figures (D), (E) and (F) are of cytosine; (G), (H) and (I) are of adenine; and (J), (K) and (L) are of guanine composition, respectively.
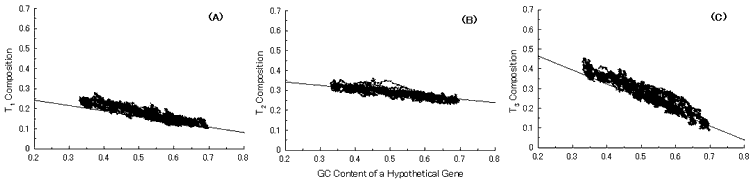
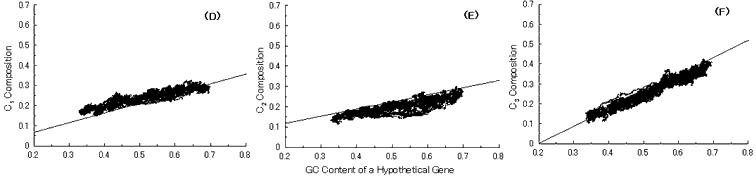
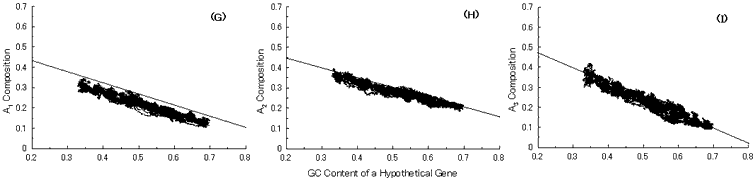
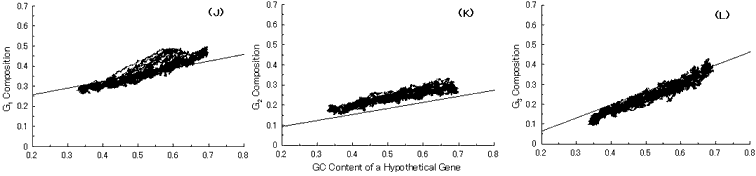
Fig. 5. Simulation of gene evolution from a GC-NSF(a) on M. tuberculosis Rv0001 gene (508 codons) with setting about 40% conserved region (200 amino acids from N-terminal end) in the ancestor protein. Linear lines in the figures represent the average values approximated with the least square method of base compositions at the codon positions of extant genes on seven microbial genomes [6]. Figures (A), (B) and (C) show the dependencies of thymine composition on GC content of a hypothetical gene at the first, second and third codon positions, respectively. Similarly, figures (D), (E) and (F) are of cytosine; (G), (H) and (I) are of adenine; and (J), (K) and (L) are of guanine composition, respectively.
Next, we put into practice the gene evolution-simulation from a B. burgdorferi AT-rich gene to confirm whether the simulation can also reproduce base compositions at three codon positions of extant genes similar to the case of GC-NSF(a) hypothetical genes (Fig. 6). But, the results showed that guanine composition at first codon position of simulated genes from the AT-rich gene considerably deviates from that of extant genes, irrespective of the establishment of a conserved region (Fig. 6). Thus, these results also support the hypothesis that extant genes evolved from GC-NSF(a) ancestor genes and not from AT-rich genes.
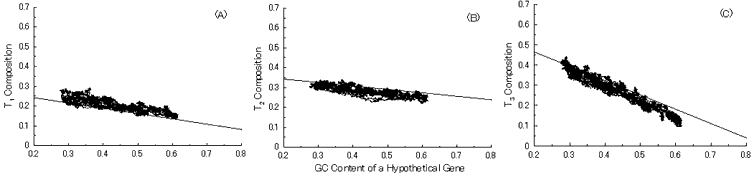
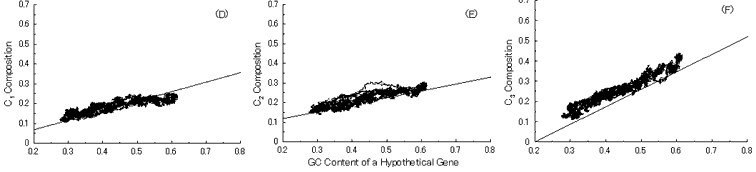
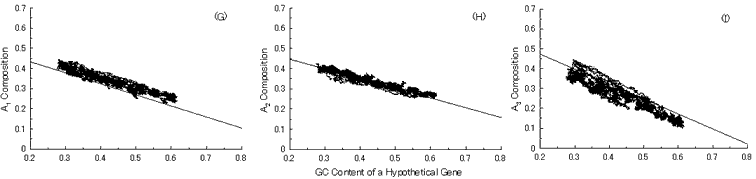
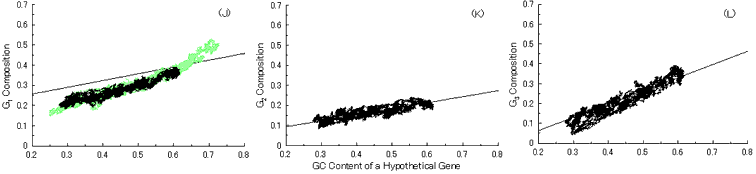
Fig. 6. Simulation of gene evolution from a B. burgdorferi AT-rich gene (BB0024; 379 codons) with setting about 40% conserved region (150 amino acids from N-terminal end) in the ancestor protein. Linear lines in the figures represent the average values approximated with the least square method of base compositions at the codon positions of extant genes on seven microbial genomes [6]. Figures (A), (B) and (C) show the dependencies of thymine composition on GC content of a hypothetical gene at the first, second and third codon positions, respectively. Similarly, figures (D), (E) and (F) are of cytosine; (G), (H) and (I) are of adenine; and (J), (K) and (L) are of guanine composition at the three base positions, respectively. In Fig. 6J, the results of simulation for guanine content at the first codon position (gray spots), which was carried out without setting any conserved region, are also shown for comparison.
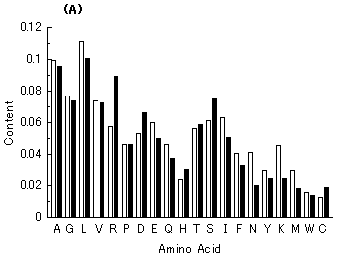
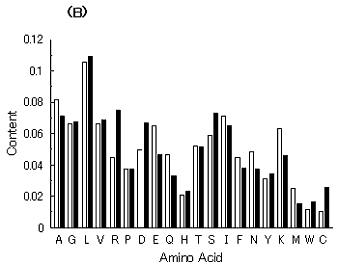
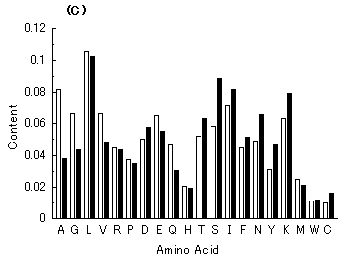
Fig. 7. Average amino acid compositions of hypothetical proteins encoded by simulated genes (closed bars) and of extant proteins (open bars). The respective amino acids are shown using one-letter symbols on the abscissa. (A) Average amino acid composition of proteins encoded by 300 simulated genes (average GC content = 50.5%) from a hypothetical GC-NSF(a) on M. tuberculosis Rv0001 with setting about 40% conserved region is compared with that of extant proteins encoded by E. coli genome (average GC content = 50.8%). (B). Average amino acid composition of proteins encoded by 300 simulated genes (average GC content = 41.1%) from a hypothetical GC-NSF(a) on M. tuberculosis Rv0001 with setting about 40% conserved region is compared with that of extant proteins encoded by H. influenzae genome (average GC content = 38.1%). (C) Average amino acid composition of proteins encoded by 300 simulated genes (average GC content = 38.7%) from a B. burgdorferi AT-rich gene (BB0024; 379 codons) with setting about 40% conserved region is compared with that of extant proteins encoded by H. influenzae genome (average GC content = 38.1%).

Fig. 8. Amino acid compositions of hypothetical proteins generated with 20 random numbers and selected with only the six conditions for formation of a water-soluble protein. In this case, the ancestor gene was not assumed, nor was the simulation of gene evolution carried out. Small dots represent amino acid compositions of the respective hypothetical proteins.
Relationship between Gene-Protein Simulation and GNC-SNS Hypothesis on the Genetic Code. We have previously provided a GNC-SNS primitive genetic code hypothesis, suggesting that the universal genetic code (64 codons; 20 amino acids) originated from GNC code (4 codons; 4 amino acids) through SNS code (16 codons; 10 amino acids) [6, 8]. Where N and S mean either of four bases and G or C, respectively. The results described in this paper may support the GNC-SNS primitive genetic code hypothesis, because both the GNC and SNS codes are GC-rich (about 83.3%) and, thus, it is supposed that primitive genes encoded by the codes must be produced as GC-rich genes like the GC-NSF(a)s and would evolve to more AT-rich genes under the universal genetic code [3, 4].
As can be seen in Fig. 8, every amino acid composition, except acidic amino acids (Asp and Glu), can be zero, suggesting that almost all amino acids has at least one substitute. This implies that the universal genetic code must be produced by duplications of a simple frame of the code, such as GNC code and SNS code. This also supports the GNC-SNS primitive genetic code hypothesis [6].
References
1. Fleischmann, R. D., et al., Science, 269, 496-512 (1995).2. For example, refer to GenomeNet in Bioinformatics center, Institute for Chemical Research, Kyoto University, Japan. Net address is http://www.genome.ad.jp/kegg/catalog/org_list.html
3. Ikehara, K., Amada, F., Yoshida, S., Mikata, Y. and Tanaka, A., Nucl. Acids Res. 24, 4249-4255 (1996).
4. Ikehara, K., Viva Origino, 29, 66-85 (2001). (a modified English version: J. Biosci., 27, 165-186 (2002)).
5. Ikehara, K. and Okazawa, E., Nucl. Acids Res. 21, 2193-2199 (1993).
6. Ikehara, K., Omori, Y., Arai, R. and Hirose, A., J. Mol. Evol., 54, 530-538 (2002).
7. Berg, J. M., Tymoczko, J. L. and Stryer, L., メBiochemistry, 5th ed. W. H. Freeman, New York 8. Ikehara, K. and Yoshida, S., Viva Origino 26, 301-310 (1998).